


Резюме: Синдромът на Марфан или MFS, е едно от най-честите генетични заболявания на съединителната тъкан, което се характеризира със скелетни, сърдечно-съдови и очни аномалии. Той се наследява като доминантен ген. Синдромът се причинява от ген, наречен FBN1, който кодира протеин на съединителната тъкан, наречен фибрилин-1. Честотата на заболяването е около 1 на 20 000, като продължителността на живота е силно понижена главно поради настъпването на сърдечно-съдови усложнения.
Ключови думи: Синдром на Марфан, мултидисциплинарен подход, диагностициране, ген FBN1, фибрилин-1, аортна аневризма и дисекация, контрол на заболяването.
MARFAN SYNDROME
Aleksandar Genkov Konakov
Abstract: Marfan syndrome, also called Marfan’s syndrome (MFS), one of the most common genetic disorders of connective tissue, is characterised by skeletal, cardiovascular and ocular abnormalities. It is inherited as a dominant trait. It is carried by a gene called FBN1, which encodes a connective protein called fibrillin-1.The incidence of the disease is about 1 in 20,000, with life expectancy severely reduced because of cardiovascular complications. As the underlying defects are unknown, MFS diagnosis is based solely on clinical criteria.
Key words: Marfan syndrome, multidisciplinary approach, diagnosis, FBN1 gene, fibrillin-1, aortic aneurysm and dissection, disease management.
Синдромът на Марфан (на латински език: Marfan syndromum) е мултисистемно нарушение на съединителната тъкан със значителна заболеваемост и преждевременна смъртност с най-чести прояви в сърдечно-съдовата, скелетната и очната система. Той е открит и описан за първи път през 1896 г. от парижкия професор по педиатрия Антоан Бернар Жан Марфан (1858-1942 г.), който съобщава за необичайно дългите и тънки крайници и пръсти на ръцете и краката и за други скелетни аномалии, констатирани при прегледа на 5-годишното момиченце Габриел. Ето защо и до днес синдромът се нарича на негово име.
Генетичното заболяване се предава по автозомно-доминантен път и засяга с еднаква честота и двата пола, като не показва расови различия или географски предпочитания. Честотата на класическия синдром на Mарфан е около 2-3 на всеки 10 000 души. Около една четвърт от засегнатите индивиди нямат фамилна анамнеза за заболяването, което показва, че в някои случаи се касае за нововъзникнали мутации. Увреждането се локализира в съставката колаген в съединителната тъкан. Заболяването се дължи на различни молекулни дефекти в ген FBN1, кодиращ образуването на фибрилина. Този ген е локализиран в хромозома 15q21.1. Фибрилинът е гликопротеин, секретиран от фиброобластите, който сам или в комбинация с различни протеини образува микрофибриларна екстрацелуларна мрежа в съединителната тъкан. Диагнозата се поставя на базата на характерните симптоми и белези на заболяването и се потвърждава с молекулярно-биологични изследвания за откриване на мутации в FBN1 гена.
Фигура 1
Съвкупността от дълги и тънки крайници, дислокация на очната леща и дефекти на клапите на сърцето и аортата обикновено са достатъчни, за да се постави диагноза синдром на Марфан с достатъчна увереност. Има повече от тридесет други клинични характеристики, които в различна степен са свързани с този синдром, включващи най-вече поражения по скелета, кожата и ставите. Интересно е, че интелектът при пациентите със синдром на Марфан е напълно съхранен и не се засяга от това тежко заболяване.

Фигура 2
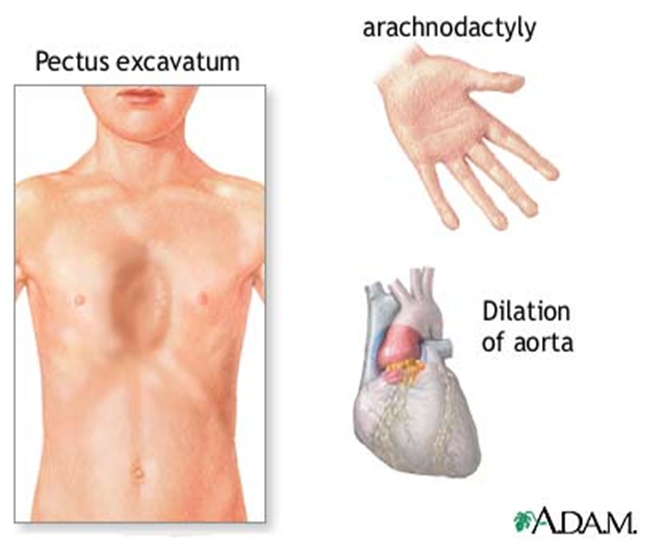
Повечето от лесно видимите признаци са свързани с скелетната система и опорно-двигателния апарат. Много хора със синдром на Марфан имат много по-висок ръст от средната височина (Фиг.1). Повечето от тях имат издължени тънки крайници с дълги и тънки пръсти на ръцете и краката (Фиг.2), специфични състояния познати на медицината като долихостеномелия и съответно арахнодактилия (Фиг.3). Ръцете на отделния индивид могат да бъдат непропорционално дълги с тънки и слаби китки. В допълнение към много високия ръст и абнормалните пропорции на крайниците, синдрома на Марфан може да доведе и до други скелетни аномалии, като необичайно изкривяване на гръбначния стълб (т.нар. сколиоза), патологични вдлъбнатина (pectus excavatum) или издатина (pectus carinatum) на гръдната кост (Фиг. 3). Други симптоми са необичайна гъвкавост на ставите, високо небце, плоскостъпие, екстензия на лакътните стави, наведени рамене, необясними стрии по кожата, както и някой лицеви аномалии – енофталмия, долихоцефалия, ретрогнатия. Синдромът може също да причини и болки в ставите, костите и мускулите при някои пациенти. Някои хора с Марфан имат говорни нарушения, произтичащи от симптоматично високото небце и малките челюстите. Понякога може да се появи и ранен остеоартрит.
Фигура 3
Синдромът на Марфан се отразява много сериозно върху очите и зрението, като най-честите офталмологични прояви са късогледството и астигматизмът, но е възможно развитие и на далекогледство. Други характерни симптоми са сублуксацията (разместването) на кристалната леща в едното или двете очи, което води до вродена катаракта и ектопия на очната леща (еctopia lentis) при 80% от пациентите. Второстепенни прояви на синдрома са плоската роговица, удължената очна ос, хипопластичния ирис или хипопластичния цилиарен мускул, причиняващ миоза. Някои от тези аномалии могат да бъдат открити при обикновен прегледи от офталмолог или оптиометрист, а за други се изискват специализирани изследвания и апаратура. Понякога проблемите с очите и зрението се появяват впоследствие, след отслабването на съединителната тъкан, което може да доведе дори до отлепване на ретината. Ранната появата на глаукома може да бъде друг проблем, свързан със синдрома на Марфан.
Най-сериозните признаци и симптоми, свързани със синдрома на Марфан включват сърдечно-съдовата система. Безпричинна умора, задух, сърцебиене, високо кръвно или ангина пекторис са само някои от множеството прояви на заболяването. Студените ръце, длани и стъпала също могат да бъдат обяснени и свързани със синдрома, защото са последица от нарушеното кръвообращение и циркулация на кръвта. Сърдечните шумове, отклоненията в ЕКГ, или симптомите на стенокардия също могат да манифестират синдром на Марфан. Бременните със синдрома на Марфан могат да получат разкъсване на аортата (т.нар. аортна дисекация), което в повечето случаи е фатално. Ето защо като профилактична мярка се прилага ехокардиография на всеки 6 до 10 седмици от бременността. Симптомите на регургитация от пролапс на митралната клапа (т.е. хлабави и дълги хорди – залавни сухожилия) или аортната клапа (които клапи контролират потока на кръвта през сърцето) са друга животозастрашаваща последица, свързана със синдром на Марфан. Освен това, основните признаци, които биха накарали болния да отиде на лекар, който да свърже сърдечно-съдовите му оплаквания със синдрома е разширението на аортната клапа или аортната аневризма, което е резултат от разрушаването на еластичната мрежа в нейната стена (Фиг. 3). Засягането на аортата при синдрома на Марфан може да доведе до разкъсването на този най-голям кръвоносен съд в организма, което влече след себе си изключително тежки последици – от масивен кръвоизлив до летален изход.
Пациентите със синдром на Марфан са предразположени към развитие на спонтанен пневмоторакс. При спонтанен едностранен пневмоторакс, въздуха излиза от белите дробове и навлиза в плевралната кухина, между белите дробове и гръдния кош. Белите дробове са притиснати и могат да колабират. Последното може да стане причина за развитие на цианоза, болка, повърхностно дишане, а ако тези усложнения не се лекува – това може да доведе дори и до смърт. Синдромът на Марфан е асоцииран още със сънната апнея и идиопатичната обструктивна белодробна болест.
За съжаление, това тежко заболяване е неизлечимо, а терапията, която се прилага е само симптоматична и цели облекчаване на моментното състоянието на пациента и забавяне развитието на сърдечно-съдовите аномалии. Стандартна фармакологична терапия на синдрома по отношение на сърдечната недостатъчност и свързаните с нея усложнения е приложението на бета-адренергични блокери, които намаляват или профилактират аортната аневризма и дисекация, тъй като редуцират стреса на стената на възходящата аорта. Ползата им се дължи и на отрицателния инотропен и хронотропен ефект. При неуспех от медикаментозното лечение, се предприема хирургична интервенция. При болни с митрален клапен пролапс, митрална регургитация и аортна регургитация се препоръчва антибиотична профилактика с цел превенция от ендокардит. Препопъчва се също и редовен контрол на кръвното налягане и пулса на болните със синдром на Марфан и поддържането им в ниски стойности, тъй като високото кръвно (хипертонията) е основната причина за формирането на аортната аневризма, разкъсването на която може да бъде фатално също както при разрива на сърце.

Фигура 4
При случаите с повишен риск за отлепване на ретината и глаукома, се изисква проследяване от офталмолог и имплантация на изкуствени лещи или хирургична афакия след достигане на определена възраст. Деформациите на костната система (кифоза и сколиоза) изискват контрол от ортопед, а в някои случаи – и хирургична намеса и интервенция.
Интересен факт е, че според канадският египтолог Алвин Бъридж (Alwyn Burridge) първото изображение на човек, който е страдал от синдрома на Марфан e египетският фараон Аменхотеп IV (Ехнатон), живял през периода 1424-1388 г. пр. Хр., който е син на Аменхотеп III. Следователно синдромът не е ново заболяване, а такова съществуващо от преди хиляди години.
Аменхотеп IV Ехнатон (Фиг. 4) е известен като “фараона-революционер”, тъй като той е променил религията на древен Египет – от политеизъм, т.е. от вяра в много богове в монотеизъм, т.е. вяра само в един бог – Атон или бога Слънце, но също така е останал в историята и като съпруг на красавицата Нефертити. Освен с религиозната реформа, Аменхотеп IV Ехнатон е известен и с новаторството си в изобразителното изкуство – за първи път по неговото време фараонът и неговото семейство са рисувани като живи хора.

Фигура 5
Благодарение на останалите изображения на главата и пръстите на Аменхотеп IV Ехнатон, които свидетелстват за долихоцефалия и арахнодактилия, както и многобройните скулптури, показващи неговата хабитуална структура (висок ръст и долихостеномелия), учените смятат, че той е страдал от синдрома на Марфан, чиито усложнения вероятно са довели до ранната му и преждевременна смърт.
Други известни исторически личности, при които също е поставена диагнозата синдром на Марфан, са италианския композитор и виртуозен цигулар Николо Паганини (1782 – 1840 год.), както и шестнадесетият президент на САЩ и първият президент от Републиканската партия – Абрахам Линкълн (1809 – 1865 год.). Спекулира се, че с това генетично нарушение е бил и Осама Бин Ладен (1957 – 2011 год.) – основателят на терористичната организацията Ал Кайда, отговорна за Атентатите от 11 септември 2001 г. в САЩ!
Библиография/ References
- Judge D., Dietz H. Marfan’s syndrome. Lancet 2005; 366: 1965-76;
- Ho N., Tran J., Bektas A. Marfan’s syndrome. Lancet 2005; 366: 1978-1981;
- Marfan MA-B. Un cas de deformation congenitale des quatre membres, plus prononcee aux extremites, caracterisee par l’allongement des os avec un certain degre d’amincissement. Bull Mem Soc Med Hop Paris 1896;13:220–6;
- Pyeritz RE. Marfan syndrome and other disorders of fibrillin. In: Rimoin DL, Connor JM, Pyeritz RE, eds. Principles and Practice of Medical Genetics. 3rd edn. New York: Churchill Livingstone, 1997:1027–66;
- Murdoch JL, Walker BA, Halpern BL, Kuzma JW, McKusick VA. Life expectancy and causes of death in the Marfan syndrome. N Engl J Med 1972;286:804–8;
- Motro M, Fisman EZ, Tenenbaum A. Cardiovascilar management of Marfan syndrome. IMAJ 2008;10:182–5;
- Nahum Y, Spierer A. Ocular features of Marfan syndrome: diagnosis and managment. IMAJ 2008;10:179–81;
- Avivi E, Arzi H. Paz L, Caspi I, Chechik A. Skeletal manifestations of Marfan syndrome. IMAJ 2008; 10:186–8;
- Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation 2004;109:2807–16;
- De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet 1996;62:417–26;
- http://www.marfan.org/marfan/ – National Marfan Foundation;
- http://www.ima.org.il/imaj/
Снимки и схеми – http://www.marfanvic.org.au
